What is phylogeny
Phylogenetics is the analysis of the evolutionary relationships among species. The evolutionary relationship between these species can be portrayed visually through a phylogenetic tree. Through the analysis of DNA and protein sequences, phylogenetics can elucidate the relation between species and their features of interest such as a gene, protein, or phenotype [1]. By studying how each species has diverged from one another, and analyzing the characteristics that have remained conserved, we can use phylogenetics to explore the evolution of genes and the biological similarities among pertinent species [2].
How to Construct a Phylogenetic Tree
A phylogenetic tree is a great way to visually present the evolutionary relationship for a gene or protein among pertinent species. The first step in creating a phylogenetic tree is to collect the DNA/RNA/Protein sequences of the gene of interest for all of the species you would like to study [3]. After collecting the sequences, they must be aligned, and this can be done through using software programs like ClustalOmega or Mega.
There are multiple ways you can construct a phylogenetic tree. One particular tree that you can construct is a maximum likelihood tree. This phylogenetic tree is constructed based on the most likely path of divergence based on the sequences that were aligned. So, the order in which a species branches from the phylogenetic tree is based on the most likely chances of something diverging at that time [3]. A maximum likelihood phylogenetic tree can be generated by Mega or ClustalOmega.
Another possible phylogenetic tree that you can construct is a neighbor joining tree. The neighbor joining tree is determined by the differences in the aligned sequences among two species [4]. This analysis is conducted for every species and then a phylogenetic matrix is formed. The closer the "neighbor" or species, the smaller the distance is on the neighbor joining tree [4]. The neighbor joining tree can also be generated by Mega or ClustalOmega.
There are multiple ways you can construct a phylogenetic tree. One particular tree that you can construct is a maximum likelihood tree. This phylogenetic tree is constructed based on the most likely path of divergence based on the sequences that were aligned. So, the order in which a species branches from the phylogenetic tree is based on the most likely chances of something diverging at that time [3]. A maximum likelihood phylogenetic tree can be generated by Mega or ClustalOmega.
Another possible phylogenetic tree that you can construct is a neighbor joining tree. The neighbor joining tree is determined by the differences in the aligned sequences among two species [4]. This analysis is conducted for every species and then a phylogenetic matrix is formed. The closer the "neighbor" or species, the smaller the distance is on the neighbor joining tree [4]. The neighbor joining tree can also be generated by Mega or ClustalOmega.
Maximum Likelihood Phylogenetic Tree for DYRK1A
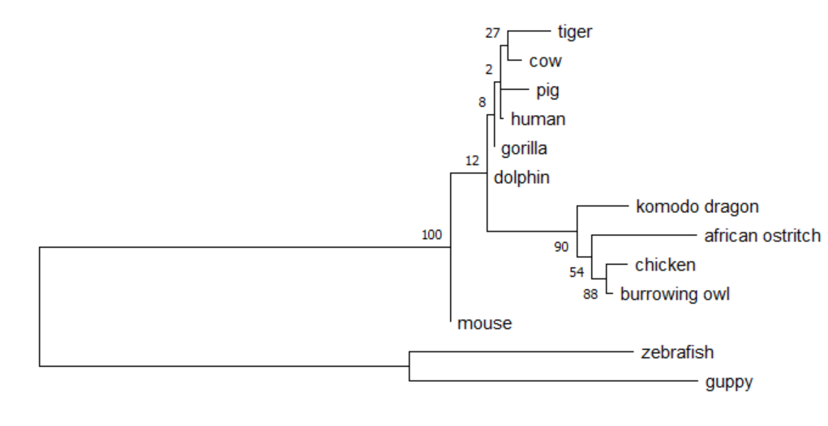
Discussion
Based on the maximum likelihood phylogenetic tree for DYRK1A, it is evident that DYRK1A is an ancient gene and its homologs have a robust evolutionary relationship. Additionally, it is clear that the organisms with very diverse and complex nervous systems show high similarity for this important regulatory gene because of the branching of the mammals (ex. human, pig, gorilla, dolphin). Although zebrafish is not a mammal, it is one of the first to diverge from this phylogenetic tree which suggests that the basic neural developmental pathways present in zebrafish will also be present in humans.
References
1. Phylogenetics: An introduction. Retrieved from https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
2. Yang Z, Rannala B. (March 2012). Molecular phylogenetics: principles and practice. Retrieved from https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
3. ClustalOmega: https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20190308-024243-0917-65706474-p2m&showColors=true&tool=clustalo
4. "MEGAX-Help". Megasoftware.Net, 2020, https://www.megasoftware.net/web_help_10/index.htm#t=First_Time_User.htm.
2. Yang Z, Rannala B. (March 2012). Molecular phylogenetics: principles and practice. Retrieved from https://www-nature-com.ezproxy.library.wisc.edu/articles/nrg3186
3. ClustalOmega: https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20190308-024243-0917-65706474-p2m&showColors=true&tool=clustalo
4. "MEGAX-Help". Megasoftware.Net, 2020, https://www.megasoftware.net/web_help_10/index.htm#t=First_Time_User.htm.
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
